Терапия антителами дает надежду пациентам с прионными заболеваниями
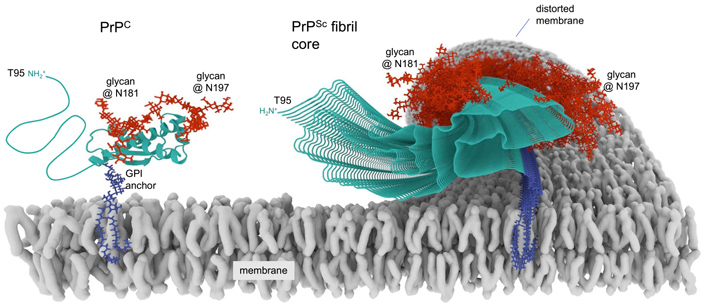

Рис. 1. Нормальная (PrPC) и прионная (PrPSc) форма белка PrP на клеточной мембране (membrane). Белок PrPC в норме — мембранный белок, который заякорен в клеточной мембране c помощью гликозилфосфатидилинозитола (GPI (glicosylphosphatidylinositol) anchor, показан фиолетовым). Бирюзовым цветом изображен сам белок, состоящий из трех альфа-спиралей и двух бета-листов. Красным цветом обозначены остатки сахаров, модифицирующие белок. При неправильной укладке в прионную форму PrPSc образуют фибриллярную структуру (PrPSc fibril core), состоящую из бета-листов. Аномальное расположение сахаров приводит к изменению структуры мембраны (distorted membrane). Накопление PrP приводит к гибели клеток и неизлечимым нейродегенеративным заболеваниям. Иллюстрация из статьи A. Kraus et al., 2021. High-resolution structure and strain comparison of infectious mammalian prions
Болезнь Крейтцфельдта — Якоба (БКЯ) — это тяжелое, хотя и редкое, нейродегенеративное заболевание. Оно возникает из-за накопления в коре и других отделах головного мозга прионов, представленных аномальной формой мембранного белка PrP. Устранить эти прионные агрегаты невозможно. Болезнь быстро прогрессирует, и большинство заболевших умирает в течение 8–26 месяцев. Тем не менее попытки найти препарат, который сможет хотя бы замедлить течение БКЯ, не прекращаются. Ученым из Британского института прионных болезней удалось провести первое исследование действия антител к белку PrP на пациентах с БКЯ. Связываясь с белком PrP, антитела тормозят образование прионных бляшек. У некоторых пациентов действительно отмечалась некоторая задержка развития симптомов. Однако полноценные выводы об эффективности этого подхода к лечению БКЯ можно будет сделать только после проведения более крупных исследований.
Многие нейродегенеративные заболевания сопровождаются накоплением аномальных форм белков — так называемых амилоидных агрегатов. При болезнях Альцгеймера, Паркинсона, Гентингтона образуются плотные нерастворимые бляшки белков (Aβ-пептидов и тау-белка, α-синуклеина, гентингтина, соответственно). Они возникают из-за особого способа укладки белковых молекул (см. статью Инфекционность амилоидов). Накопление таких агрегатов нарушает функцию нейронов и приводит к когнитивным, двигательным и другим неврологическим нарушениям.
Однако среди всех амилоидных заболеваний выделяется особая группа — прионные болезни. К ним относятся болезнь Крейтцфельдта — Якоба (БКЯ), куру, синдром Герстмана — Штраусслера — Шейнкера, фатальная семейная бессонница. При всех этих заболеваниях нарушается работа нервной системы, болезнь неуклонно прогрессирует, и пациенты умирают. Различаются эти заболевания в основном вариантами симптомов и скоростью наступления смерти. Например, средняя длительность БКЯ — приблизительно год с проявления симптомов (нарушение внимания и памяти, переходящие в деменцию и неспособность двигаться). В то же время, фатальная семейная бессонница длится (с момента наступления у пациента тяжелой бессонницы до его смерти) до нескольких лет.
Все эти заболевания связаны с одним-единственным белком — PrP (он кодируется геном PRNP), который в норме присутствует на поверхности клеток нервной системы. Функция его изучена не очень глубоко, но считается, что он участвует в регуляции сна и памяти, а также в развитии нервных связей в мозжечке (см. обзор M. Wulf et al., 2017. The biological function of the cellular prion protein: an update). Но в отличие от многих других белков у него есть одна неприятная особенность: некоторые мутации приводят к тому, что он превращается не просто в амилоидный белок, который образует бляшки, а в настоящий инфекционный патоген.
Болезнь Крейтцфельдта — Якоба — самая распространенная из прионных болезней человека. В большинстве случаев она возникает спонтанно из-за нарушения фолдинга белка PrP. В норме белок из последовательности соединенных в нужном порядке аминокислот (первичной структуры) приобретает свою третичную (трехмерную) структуру, в результате чего белок и становится способным выполнять свои функции. Как и любой биологический процесс, фолдинг не всегда проходит безошибочно. Однако такие нарушения контролируются белками-шаперонами, а бракованные белки разрушаются с помощью специальных клеточных органелл — протеасом и лизосом. Если эти системы не работают, то неправильно сложенный белок может превратиться в будущее звено амилоидной структуры: сотни и тысячи молекул неправильно сложенного белка будут накапливаться с каждым следующим нарушением фолдинга.
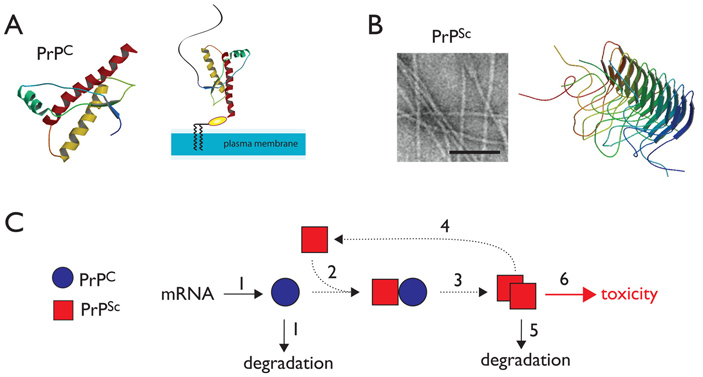

Рис. 2. A — клеточная форма белка PrP (PrPC), закрепленная в плазматической мембране (plasma membrane) клетки. B — аномальная форма белка PrPSc собирается в фибриллярные агрегаты. Слева — электронная микрофотография фибрилл PrPSc (длина масштабного отрезка — 1000 Å). Справа — одна из моделей укладки прионных белков. C — упрощенная модель распространения PrPSc (красный). МРНК гена PRPN транслируется в клеточную форму белка (PrPC), которая также может подвергнуться деградации; затем белок PrPC связывается с аномальной формой PrPSc (2) и сам переходит в аномальную форму (3). Некоторые молекулы PrPSc становятся затравкой для нового перехода (4). Некоторые агрегаты PrPSc разрушаются системами контроля качества белков (5), но в случае их длительного присутствия и накопления агрегаты становятся токсичными для клеток. Рисунок из статьи S. Ghaemmaghami et al., 2014. Successes and Challenges in Phenotype-Based Lead Discovery for Prion Diseases
Если этот белок — PrP, то ситуация выглядит немного по-другому. Неправильно сложенный PrP (его часто обозначают PrPSc, где буквы Sc — аббревиатура от scrapie, английского названия почесухи овец, которая также имеет прионную природу) не просто накапливается за пределами клетки, а заставляет другие белки PrP на ее поверхности превращаться в неправильную форму PrPSc. Эта форма также способна передаваться от клетки к клетке, распространяясь по нервной системе. Молекулярный механизм перехода в патогенную форму еще изучается. Известно, что для него нужен прямой контакт между нормальной и прионной формой белка, а в процессе перехода не затрачивается энергия (в отличие от нормального фолдинга белков). Всё это можно сравнить со сработавшей мышеловкой в комнате, наполненной тысячами заряженных мышеловок. Срабатывание каждой мышеловки влечет за собой срабатывание соседних с ней.
В 10–15% случаев БКЯ имеет генетическую природу. Мутации в гене PRNP приводят к неправильному фолдингу белка. Однако присутствие таких мутаций не всегда приводит к болезни. Как и в спонтанной форме, дефектный белок должен сначала накопиться перед тем, как вызвать заболевание. Спонтанная формы БКЯ возникает обычно в пожилом возрасте, когда клеточные системы контроля качества белков менее эффективны. Генетическая же (семейная) форма заболевания возникает раньше — в 30–50 лет.
Еще две формы БКЯ относятся к случаям, когда PrPSc становится самым настоящим инфекционным агентом. Ятрогенная форма БКЯ возникает, если прион заносится в организм человека во время медицинских вмешательств. Исторически самая распространенная причина — использование соматотропина (гормона роста), который до разработки рекомбинантного белка получали из гипофиза погибших людей. Также передача может произойти из-за пересадки роговицы или твердой мозговой оболочки пациента — носителя прионов, либо из-за недостаточной стерилизации операционных инструментов. Избавиться от прионов можно только нагреванием предметов до 500°C в течение долгого времени (по сути, сжигая любой биологический материал) либо комбинацией длительного вымачивания в щелочи или хлорке и нагревания под давлением в дополнение к уже используемым методам дезинфекции (см. рекомендации ВОЗ). Причина такой устойчивости — в особой трехмерной структуре прионов. У большинства белков, в том числе и PrPC, только часть структуры состоит из бета-листов. У PrPSc же почти половина белка уложена в виде этой конфигурации.
Прионы могут передаваться от одного вида животных к другому. Новый вариант БКЯ (см. Variant Creutzfelder — Jacob disease) зафиксирован после эпизоотии губчатой энцефалопатии крупного рогатого скота, которая более известна как «коровье бешенство». Болезнь развивалась в основном у людей, которые ели мясо зараженных коров. В мышцах нет прионов, однако аномальный белок есть в остатках нервной ткани. Так как прионы помимо нагревания устойчивы к протеазам (ферментам, разрушающим белки), они поглощаются клетками кишечника, а затем переносятся в лимфатическую систему, которая взаимодействует с нервной (см. S. Ghosh, 2004. Mechanism of intestinal entry of infectious prion protein in the pathogenesis of variant Creutzfeldt–Jakob disease). Также новый вариант БКЯ в очень редких случаях может передаваться от человека к человеку. Зарегистрированы четыре случая передачи через донорскую кровь, поэтому в большинстве стран приняты меры, ограничивающие жителей стран в донорстве во время эпизоотии «коровьего бешенства». Важно отметить, что БКЯ не переносится воздушно-капельным путем или при бытовом контакте.
Каждый год БКЯ регистрируют в среднем у одного человека на миллион, то есть количество таких пациентов всё равно значительно. От болезни нет лечения, но это не значит, что ученые оставили попытки его разработать. К счастью, болезнь можно смоделировать на животных. Если, например, трансгенных мышей заразить человеческими прионами, то в течение времени у них развиваются симптомы БКЯ. Некоторые из таких исследований уже дали результаты и перешли в стадию клинических испытаний. Например, была попытка приспособить для лечения БКЯ уже известные препараты: флупиртин (анальгетик), мепакрин (антипаразитарное средство) и доксициклин (антибиотик). В культурах клеток и у мышей эти препараты смогли снизить количество прионов. Однако в рандомизированных клинических исследованиях они не показали значительных результатов.
Причин этому может быть несколько: во-первых, сложно набрать подходящую группу пациентов. Болезнь может развиться в любом возрасте и прогрессировать с разной интенсивностью. Как и в случае других нейродегенеративных заболеваний (например, болезни Альцгеймера) накопление амилоидов может происходить годами, а симптомы проявляются уже при их достаточном накопленном уровне. Пациенты с БКЯ умирают в течение примерно года с появления симптомов. В реальной жизни две трети этого времени уходит только на установление диагноза. Вторая причина — препараты могут быть эффективными против накопления прионов у животных, но не против прионов человека. Третья — препарат должен попасть в головной мозг, который отделен от кровотока гематоэнцефалическим барьером (ГЭБ) (подробнее о проблемах этих исследований см. в обзоре S. M. Vallabh et al., 2020. Towards a treatment for genetic prion disease: trials and biomarkers).
Еще один возможный способ борьбы с образованием прионных бляшек — использование антител к белку PrPC. Антитела связываются с белком и не дают ему возможность взаимодействовать с аномальной формой PrPSc, образовывать бляшки и способствовать развитию болезни. Такой подход был предложен еще в 2001 году лабораторией Стэнли Прузинера — первооткрывателя прионов (D. Peretz et al., 2001. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity). Однако до настоящего времени он не применялся на человеке. Группа исследователей из Британского института прионных болезней (Institute of Prion Diseases) решила закрыть этот пробел. Результаты исследования опубликованы в свежем номере журнала The Lancet Neurology.
Сперва исследователи создали антитела к подходящему участку человеческого белка PrP. Как и предполагалось, в культуре клеток и у зараженных мышей антитела связывались с прионами и не давали образовываться новым бляшкам. Кстати, эти антитела не были зарегистрированы как отдельный лекарственный препарат — они получили специальную лицензию для использования в этой программе. Строго говоря, это исследование не было классическим клиническим исследованием: авторам удалось набрать только шесть пациентов, а этого недостаточно, чтобы клиническое исследование имело статистическую значимость. Критерием для отбора пациентов были диагностированная БКЯ или серьезное подозрение на БКЯ не на последних стадиях, а также наличие у пациентов возможности приезжать в Лондон за лечением. Кроме того, из-за деликатности и строгости исследования команда ученых не могла принять сразу много пациентов. Пациенты начинали терапию по одному, а авторы могли одновременно работать только с тремя пациентами.
В исследовании приняли участие пять пациентов со спонтанной формой БКЯ и один с ятрогенной формой (ставшей результатом лечения соматостатином, полученным из гипофиза зараженных людей). После тщательного обследования (МРТ, анализ спинномозговой жидкости и функций нервной системы) пациентам внутривенно вводили антитела. Начальная доза составляла 1 мг/кг, затем ее увеличивали — сперва до 10 мг/кг, а потом до 80 мг/кг. В такой дозировке антитела вводили пациентам каждые две недели. Лечение прекращалось либо со смертью пациента, либо если у него проявлялась настолько серьезная побочная реакция, что он или его опекуны решали выйти из исследования.
При каждой инъекции у пациентов оценивали состояние с помощью шкалы развития прионных заболеваний, которую разработали в Институте прионных болезней. Она отражает двадцать параметров, охватывающих когнитивную функцию, речь, подвижность, способность к уходу за собой и питанию, а также контроль за функцией мочевого пузыря и кишечника. Чтобы понять, как у пациентов прогрессирует заболевание, течение БКЯ у них сравнивали с данными от пациентов такого же возраста со схожим происхождением и течением болезни. К сожалению, формальный статистический анализ, который обычно проводят в таких исследованиях, не давал никаких значимых различий между пациентами. Поэтому оценить эффективность можно только в сравнении с похожими случаями болезни (рис. 3). Если пациент дольше сохранял высокое значение по шкале развития прионных заболеваний, чем нелеченый пациент, это уже считалось успехом.
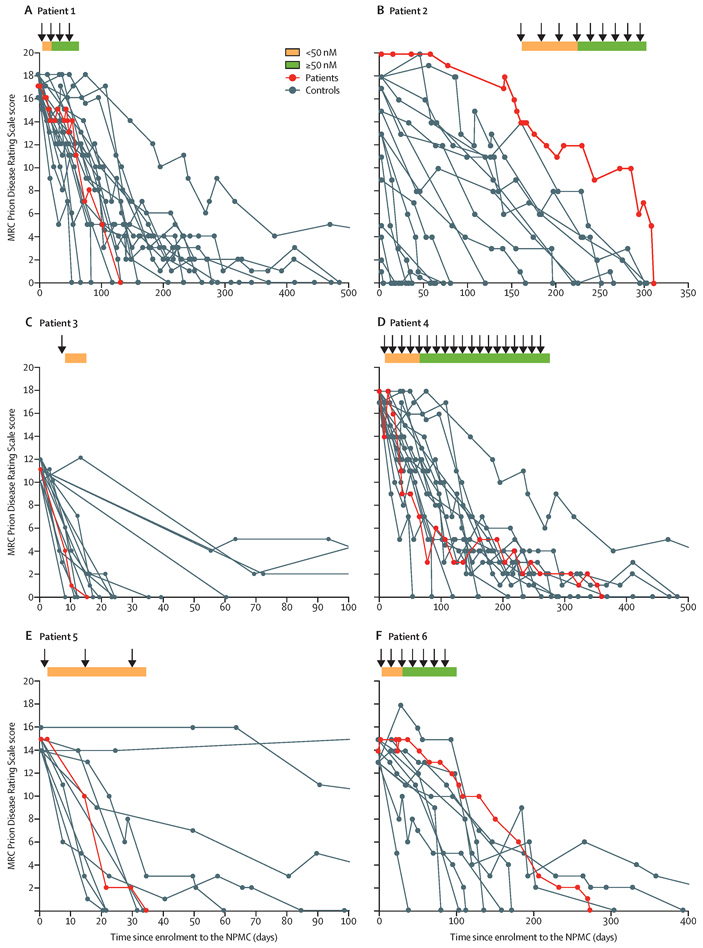
Рис. 3. Показатели по шкале развития прионных заболеваний (MRC Prion Disease Rating Scale Score) для шести пациентов, участвовавших в обсуждаемом исследовании. Красным цветом выделены траектории показателей пациентов, серым — больных из контрольной группы из национальной когорты мониторинга прионных заболеваний (NPMC, National Prion Monitoring Cohort). Стрелками обозначено введение препарата. Желтым обозначен период, когда антитела не достигли целевой концентрации в спинномозговой жидкости, зеленым — период достигнутой концентрации. По горизонтальной шкале — время с момента попадания в NPMC, в днях. Рисунок из обсуждаемой статьи в The Lancet Neurology
Первый пациент находился в стабильном состоянии после того, как концентрация антител в спинномозговой жидкости достигла целевого значение 50 нмоль/л. Однако он не вернулся после рождественских праздников и прекратил участие в исследовании. Второй пациент тоже был в стабильном состоянии, но умерл от пневмонии; при этом она прожила дольше, чем больные из контрольной группы с ятрогенной формой БКЯ. Третий и пятый пациенты, к сожалению, скончались до того, как концентрация антител достигла целевого значения. У них болезнь прогрессировала слишком быстро несмотря на терапию. Состояние четвертого пациента стабилизировалось, хотя и на низких значениях шкалы. Он получал антитела, но в какой-то момент препарат закончился, и терапию не смогли продолжить. У шестого пациента болезнь прогрессировала медленно, но интенсивность заболевания увеличивалась. Терапия продолжалась, но препарат кончился, и он скончался от БКЯ через несколько месяцев.
Эти результаты могут показаться скромными, но на самом деле они очень важны. Пациенты в целом достаточно хорошо переносили терапию, за исключением некоторых особенностей спинномозговой жидкости: предположительно антитела влияли на проницаемость гематоэнцефалического барьера, но не вызывали значительной патологии. Кстати, несмотря на способность ГЭБ защищать головной мозг от большинства веществ, попадающих в кровь, антитела проникают в спинномозговую жидкость через сосудистое сплетение желудочков головного мозга. Из спинномозговой жидкости они проникли в различные отделы головного мозга. Это важное наблюдение, так как эффективность этого процесса обычно довольно низкая. К сожалению, проверить это можно было только после смерти пациентов.
Только два пациента или их представителя подписали согласие на вскрытие. После смерти авторы изучили, как прионные бляшки распределяются в головном мозге. Оказалось, что у второго пациента структура бляшек стала менее плотной (рис. 4) в теменной и затылочных зонах головного мозга, а также изменилась в зоне, окружающей желудочки. У третьего пациента головной мозг также был изучен, но так как пациент скончался до достижения оптимальной концентрации антител в крови, то никаких изменений в распределении прионных бляшек не наблюдалось
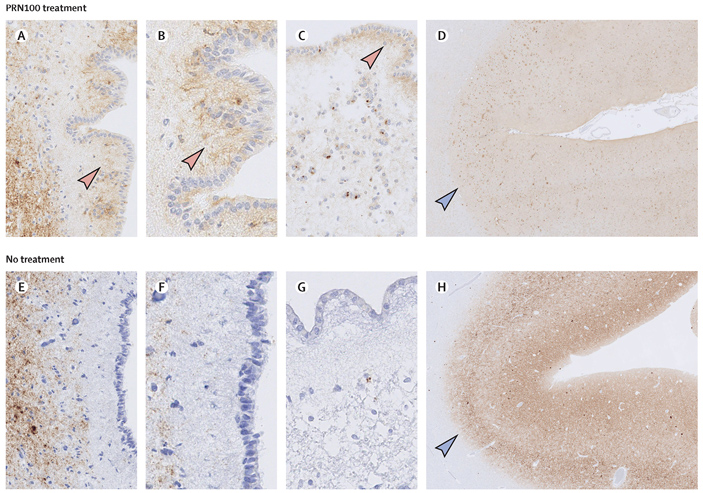

Рис. 4. Распределение прионов (коричневые пятна) в головном мозге пациента №2 (A–D) и больного с такой же формой БКЯ из контрольной группы (не получавшего лечение). У пациента №2 обнаружилось диффузное (а не компактное) распределение PrP в районе водопровода мозга (стрелки на изображениях A и B), а также в районе левого желудочка мозга (С). Обратите внимание на аналогичный участок головного мозга у пациента, не получившего лечение (E–G): окрашивание значительно различается. Распределение PrP в синапсах теменной коры пациента №2 (D) имеет слабое (как бы выцветшее) окрашивание по сравнению с этим же участком коры у пациента из контрольной группы (H). Синим цветом показаны морфологические структуры, которые окрашиваются стандартным красителем (гематоксилин+эозин). Рисунок из обсуждаемой статьи в The Lancet Neurology
Важно отметить, что у обоих обследованных посмертно пациентов не было никаких признаков токсичности «прионных» антител для головного мозга: не обнаружилось ни гибели клеток, ни скоплений микроглии, ни других признаков воспаления и изменения морфологии клеток. Таким образом терапия по меньшей мере безопасна.
Безусловно остается вопрос эффективности этой антительной терапии. Чтобы ее оценить, в первую очередь нужно исследование с большим количеством пациентов. Шесть человек недостаточно для полноценного статистического анализа различий, тем более что пациенты могут выйти из клинических исследований или не подписать согласие на некоторые процедуры (в том числе на вскрытие в случае смерти). Авторы предполагают, что они смогут набрать хотя бы 50 пациентов для последующих исследований.
Другой аспект — в идеале хотелось бы провести исследование на пациентах с как можно более ранними стадиями заболевания, чтобы успеть замедлить ход болезни. Однако, как уже упоминалось, диагностика БКЯ — это сложная задача. Накопление прионов начинается задолго до того, как у пациента проявляются симптомы. В этом прионные заболевания очень похожи на болезнь Альцгеймера: пока не существует маркеров, позволяющих выявлять их на ранней стадии. А когда процесс нейродегенерации начался, остановить его или хотя бы замедлить становится очень сложно. В 2021 году после долгих дебатов американская FDA одобрила препарат адуканумаб (Aducanumab) — антитело, останавливающее рост агрегатов бета-амилоида (накопление которых приводит к развитию болезни Альцгеймера). Тем не менее доказательств того, что препарат действительно улучшает когнитивные способности пациентов, еще нет, и их только предстоит получить.
Использование антител или молекул, предотвращающих образование прионных бляшек, — не единственный подход к лечению БКЯ. Прионные заболевания не развиваются, если белок PrP отсутствует вообще. Например, если заразить прионами мышей без соответствующего гена, то у них не возникнет никаких признаков заболевания. Избавиться от белка в организме можно, если использовать антисмысловые олигонуклеотиды (Antisense oligonucleotide) — фрагменты нуклеиновых кислот, которые связываются с мРНК определенного гена и не дают образовываться белковому продукту. В исследованиях на мышах такой способ показал эффективность (G. J. Raymond et al., 2019. Antisense oligonucleotides extend survival of prion-infected mice). Будет ли он эффективен у людей — вопрос открытый. С одной стороны, у нас есть пример препарата нусинерсен (известен под торговым названием «Спинраза»), который действует примерно по такому же принципу и применяется для лечения спинальной мышечной атрофии. С другой стороны, недавние попытки подобной терапии для лечения болезни Гентингтона провалились.
Источник: Simon Mead, Azadeh Khalili-Shirazi, Caroline Potter, Tzehow Mok, Akin Nihat, Harpreet Hyare, Stephanie Canning, Christian Schmidt, Tracy Campbell, Lee Darwent, Nicola Muirhead, Nicolette Ebsworth, Patrick Hextall, Madeleine Wakeling, Jacqueline Linehan, Vincenzo Libri, Bryan Williams, Zane Jaunmuktane, Sebastian Brandner, Peter Rudge and John Collinge. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt–Jakob disease: evaluation of a first-in-human treatment programme. The Lancet Neurology. 2022. DOI: 10.1016/S1474-4422(22)00082-5.
Екатерина Грачева